澳大利亞擁有一個成熟且規(guī)模龐大的公共和私人醫(yī)療保健系統(tǒng),對醫(yī)療器械的需求也十分旺盛。然而,想要在澳大利亞銷售、進(jìn)口、出口醫(yī)療器械,必須按照相關(guān)規(guī)定完成注冊審核并獲得準(zhǔn)入證書。這篇文章將詳細(xì)介紹這個過程。
一、澳大利亞的監(jiān)管法規(guī)
早在1966年,澳大利亞就通過了《醫(yī)療用品法案》,對醫(yī)療用品進(jìn)行管理。現(xiàn)在主要的管理法案是1989年的《醫(yī)療用品法案》,以及2002年頒布的《醫(yī)療器械法規(guī)》。這兩部法規(guī)的執(zhí)行和監(jiān)督由澳大利亞聯(lián)邦健康與老年護(hù)理部的下屬機(jī)構(gòu)TGA負(fù)責(zé)。因此,想要向澳大利亞銷售、進(jìn)出口醫(yī)療器械的制造商需向TGA提交市場準(zhǔn)入申請。
二、澳大利亞醫(yī)療器械的定義和分類
澳大利亞將醫(yī)療器械定義為由工具、儀器、用具或其他物品以及必要的附件或軟件組成的治療品。這些治療品主要通過非藥理學(xué)、免疫學(xué)或代謝方式達(dá)到主要作用,盡管這些方式可能輔助其作用。根據(jù)風(fēng)險等級,醫(yī)療器械被分為Ⅰ級、Ⅱa級、Ⅱb級、Ⅲ級、以及AIMD級(活性植入醫(yī)療器械)。
三、澳大利亞醫(yī)療器械的市場準(zhǔn)入
TGA將醫(yī)療器械分為三類進(jìn)行管理:豁免、備案和注冊。每種類型的醫(yī)療器械在上市銷售前都必須獲得政府的批準(zhǔn),并且符合醫(yī)療器械的基本要求。高風(fēng)險的醫(yī)療器械必須通過TGA的評估并在上市前獲得批準(zhǔn)。而低風(fēng)險的醫(yī)療器械可以由企業(yè)自行評估,只要符合質(zhì)量和安全條件就可以進(jìn)入市場。所有在澳大利亞生產(chǎn)的醫(yī)療器械必須符合GMP標(biāo)準(zhǔn),并在潔凈和無污染的環(huán)境下生產(chǎn)。
四、澳大利亞醫(yī)療器械上市流程解析
醫(yī)療器械在澳大利亞的上市流程是一項嚴(yán)謹(jǐn)而繁瑣的工作,需要滿足多個關(guān)鍵角色的職責(zé),明確醫(yī)療器械的定義及風(fēng)險等級,以及完成符合性評估和ARTG注冊。
(1)明確關(guān)鍵角色
首先,醫(yī)療器械在澳大利亞的注冊上市過程涉及兩個關(guān)鍵角色,即制造商(Manufacturer)和保薦人(Sponsor)。制造商負(fù)責(zé)醫(yī)療器械的設(shè)計、生產(chǎn)、包裝、發(fā)貨等,而保薦人是一位澳大利亞本地公民或公司,負(fù)責(zé)向TGA(治療性商品管理局)申請Sponsor注冊號,以幫助完成注冊過程。
(2)明確醫(yī)療器械的定義及風(fēng)險等級
接下來,需要明確醫(yī)療器械的定義及風(fēng)險等級。1989年的《醫(yī)療用品法案》對醫(yī)療器械的定義及要點(diǎn)如下:

在澳大利亞,符合《醫(yī)療用品法案1989》定義的醫(yī)療器械需要向TGA提交注冊申請。根據(jù)風(fēng)險程度和預(yù)期用途的不同,醫(yī)療器械被分為五類風(fēng)險等級,風(fēng)險級別越高,TGA監(jiān)管越嚴(yán)格。

(3)申請符合性評估證明
在醫(yī)療器械注冊上市之前,制造商需要實(shí)施合格的符合性評估程序,并接受TGA的審核,獲得有效的符合性評估證明。TGA認(rèn)可的符合性評估證明包括TGA Conformity Assessment Evidence、EC Certificate,以及基于EC-MRA和EFTA-MRA頒發(fā)的證書。
如果選擇TGA符合性評估證據(jù),證書申請過程如下:
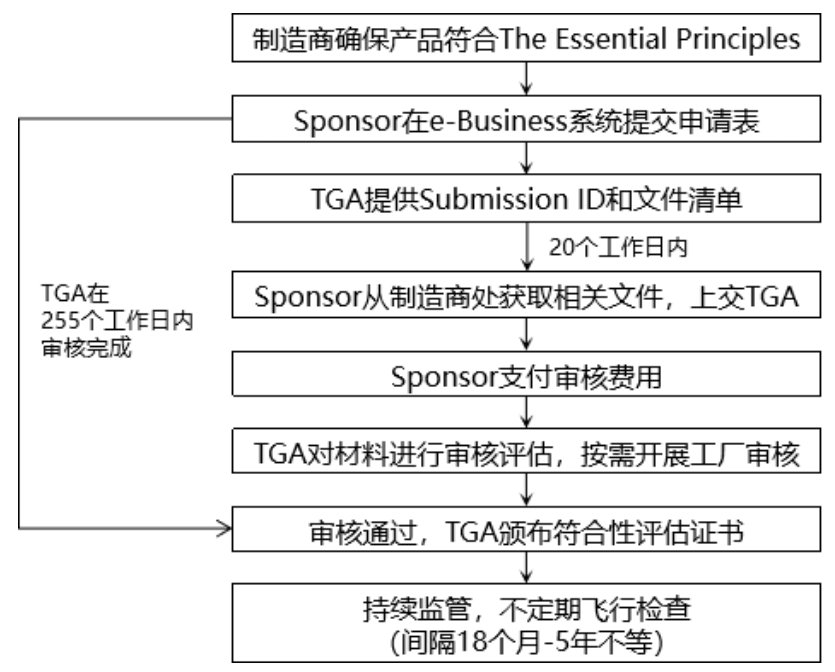
(4)提交ARTG注冊
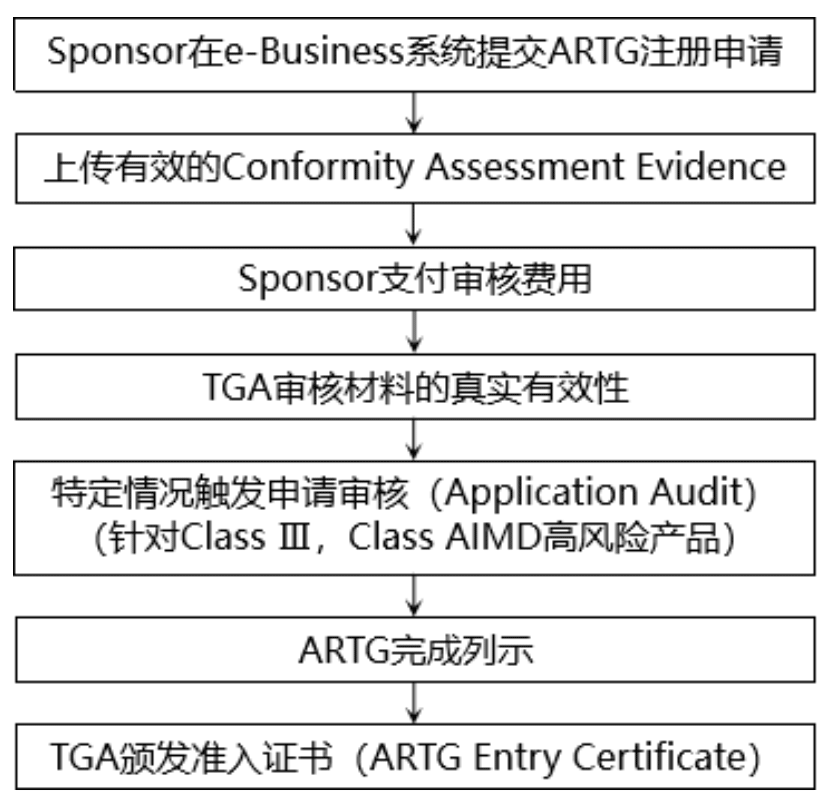
完成符合性評估后,制造商和Sponsor需要提交ARTG注冊,獲得單個醫(yī)療器械的準(zhǔn)入證書,以完成產(chǎn)品列示。只有當(dāng)醫(yī)療器械被列入澳大利亞治療性商品登記處(ARTG)后,才能在澳大利亞市場上銷售。
五、澳大利亞醫(yī)療器械的質(zhì)量體系
政府規(guī)定所有醫(yī)療器械生產(chǎn)企業(yè)的生產(chǎn)過程都必須符合與其生產(chǎn)的器械相關(guān)的質(zhì)量要求,并具備質(zhì)量保證的手段和程序。為了實(shí)現(xiàn)這一目標(biāo),澳大利亞執(zhí)行了良好制造實(shí)踐(GMP)制度,同時也向ISO 9000質(zhì)量體系標(biāo)準(zhǔn)靠攏。2017年起,澳大利亞開始實(shí)施醫(yī)療器械單一審核程序,并接受有資格的檢查機(jī)構(gòu)提供的MDSAP認(rèn)證證書。
六、澳大利亞醫(yī)療器械產(chǎn)品的市場后管理
這主要通過上市后警戒管理體系來實(shí)現(xiàn)。澳大利亞通過不良事件的調(diào)查報告、上市產(chǎn)品的實(shí)驗(yàn)室檢驗(yàn)和監(jiān)測的活動,確保所有上市后的醫(yī)療器械符合法規(guī)的規(guī)定。其上市后不良事件監(jiān)測的規(guī)定非常成熟,程序詳盡,結(jié)合了原則性和靈活性,具有強(qiáng)大的可操作性。
七、澳大利亞醫(yī)療器械產(chǎn)品的注冊變更規(guī)定
產(chǎn)品在獲得準(zhǔn)入上市后,其信息和狀態(tài)仍需持續(xù)被監(jiān)管。如果產(chǎn)品出現(xiàn)實(shí)質(zhì)性變更,制造商和Sponsor必須及時通知治療性商品管理局(TGA)。Sponsor需要登錄e-Business系統(tǒng)提交注冊變更申請。這些變更可能包括制造商、保薦人、產(chǎn)品的信息的變更,符合性評估證明信息的變更,產(chǎn)品預(yù)期用途、臨床適應(yīng)癥的變更,以及產(chǎn)品所屬種類、GMDN代碼的變更。
總的來說,要在澳大利亞銷售、進(jìn)口或出口醫(yī)療器械,必須通過一系列嚴(yán)格的準(zhǔn)入和注冊過程。這些過程保證了醫(yī)療器械的質(zhì)量、安全性和有效性,進(jìn)而保障了醫(yī)療保健系統(tǒng)的質(zhì)量。
標(biāo)簽: 出口澳洲 · 醫(yī)療器械進(jìn)出口


? 2024. All Rights Reserved. 滬ICP備2023007705號-2  滬公網(wǎng)安備31011502009912號
滬公網(wǎng)安備31011502009912號